
Introduction
Mass spectrometry (ms) is an analytical technique for measuring ion mass ratio (m/z). It identifies the composition of the sample by analysing molecular mass. Ms uses a wide range of compounds, including small organic molecules, large proteins and even complex biological molecules. This technology plays a crucial role in both qualitative and quantitative analysis, enabling scientists to determine the structure, molecular mass and chemical composition of the substance。
In mass spectrometry, the sample is ionized, then separated and the ion tested according to its m/z ratio. This information is used to generate mass spectrographs, i. E. To graphically show the relative abundance of detected ions. Each of the peaks on the mass map corresponds to a fraction of the analytical material, and the distribution of these peaks helps to identify the chemical structure and molecular mass of the compound。
Mass spectrometry is a powerful tool in many areas, including chemistry, biochemistry, pharmaceuticals and structural biology. It is often used for molecular identification, isotope analysis, study of complex mixtures and clarification of protein structures。
History of mass spectrometry
Mass spectrometry originated at the beginning of the twentieth century when the british physicist jj thomson invented a method for measuring the mass of electric particles and used it in 1913 to identify the isotopes of thiram. Thomson's technology, known as the “bracket method”, uses magnetic and electric fields to separate the ion according to the mass charge ratio. Subsequently, francis w. Aston improved thomson's work by creating the first modern mass spectrometer, which led to the discovery of many naturally occurring isotopes. Aston received the nobel prize for chemistry in 1922 because of his contribution。
In the middle of the twentieth century, technological advances, such as vacuum systems, ionizing technologies and detection methods, completely changed mass spectrometry techniques. In particular, the electronic ionizing (ei) technology invented by aj dempster in 1953 provides a more efficient molecular ionizing method. This technological advance, together with the development of the quadrapolized mass spectrometer, flight time (tof) detectors and serial mass spectrometry (ms/ms), has significantly increased the accuracy and sensitivity of mass spectrometry techniques。
The most important developments in recent decades include the inventions of electrospray ionizing (esi) and matrix-aided laser desorption/ionation (maldi), which have made it possible to analyse large biological molecules such as protein and nucleic acid. These breakthroughs have made mass spectrometry the cornerstone of the fields of proteomics, genomics and structural biology。
Mass spectrometry and instruments
Mass spectrometry works on the separation of molecules into ion based on the mass ratio, which is then tested to produce mass spectrometry. Key components of mass spectrometers include ion sources, mass analysers and detectors。
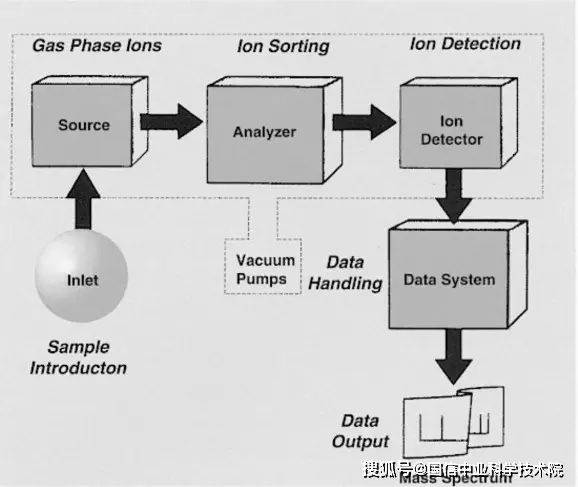
Figure 1. Components of mass spectrometers
1) ion source
The first step in mass spectrometry analysis is ionizing, converting samples into electric particles. Depending on the nature of the sample, ionizing can be achieved through various ionizing technologies. Common methods of ionizing include:
2) mass analyser
After the sample is ionized, the ion is sent to the mass analyser and separated according to its m/z ratio. The most common quality analyser types include:
3) detector
After separation by the ion mass spectrometer, it is delivered to the detector, which records the number and strength of the ion per mass ratio. Common detectors include electro-multipliers and micro-channel panels that convert ion impact into telecommunications and then process mass spectrographs。
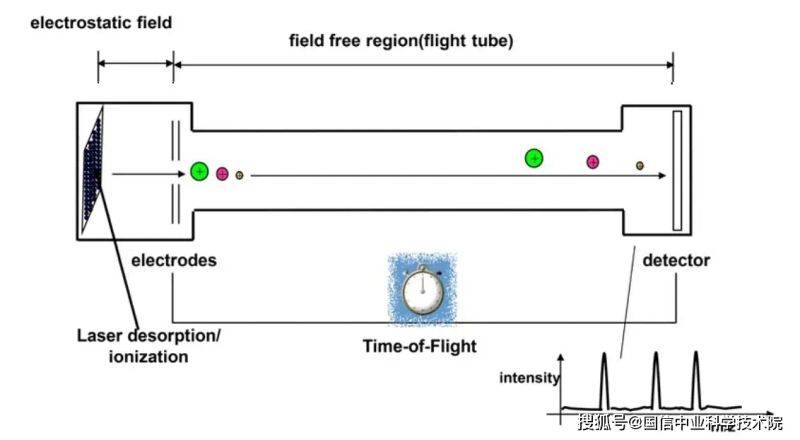
Figure 2
Iii. Separation technology and mass spectrometry
Mass spectrometry is usually used in combination with separation techniques to improve the resolution and analytical capabilities of complex mixtures. The most common combinations include:
3. 1 gas chromatography-mass spectrometry (gc-ms): in the gas chromatography-mass spectrometry (gc-ms), the gas chromatography method is used to separate volatile compounds from the boiling point in the sample before they are delivered to the mass spectrometer. This combination is well suited for analysing complex mixtures such as small organic molecules, environmental pollutants and refined oil or petrochemical products。
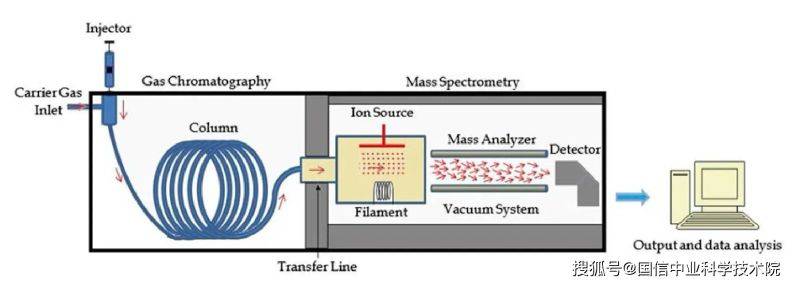
Figure 3 gc-ms system chart
3. 2 liquid chromatography-mass spectrometry (lc-ms): lc-ms are used to separate non-volatile compounds from liquids, including proteins, beryllium and medicines, before mass spectrometry is performed. Efficient liquid chromatography (hplc) is usually used in conjunction with ms to analyse complex biological samples and pharmaceutical metabolites。
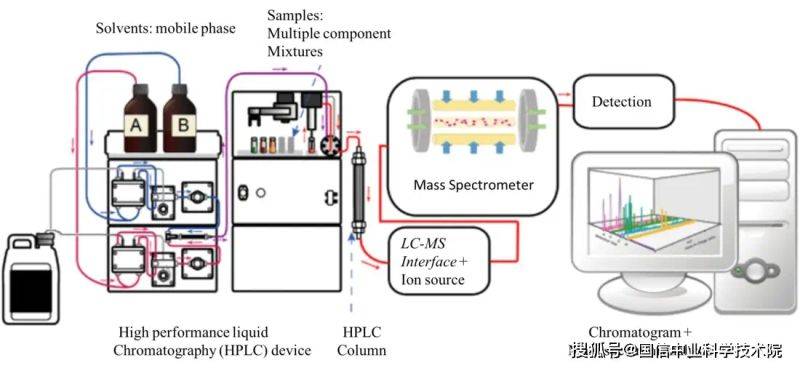
Figure 4. Lc-ms system charts
3. 3 ucps (ce-ms): ce-ms is a powerful technology that can be separated from the electric field by the size and charge of the electrical material (e. G., beryllium and nucleic acid). This method is particularly useful in proteomics and biological molecular analysis。
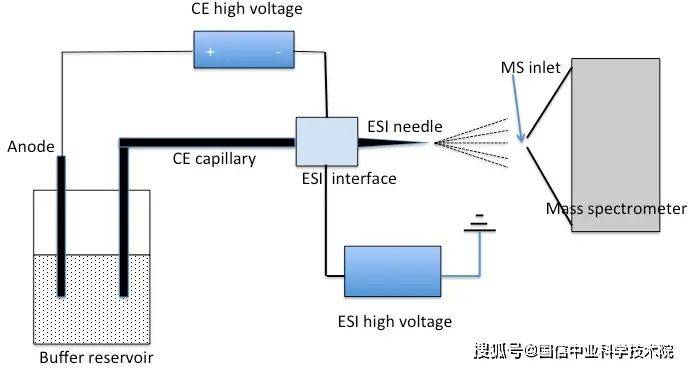
Figure 5. Ce-ms system chart
Iv. Mass spectrometry applications
Mass spectrometry has become an indispensable tool in structural biology, providing key information on the structure, dynamics and interactions of biological molecules such as proteins, nucleic acids and lipids. Some of the key applications of mass spectrometry in structural biology include:
Protein identification and presentation: mass spectrometry is widely applied in proteomics, and proteins are identified and displayed through analysis of pelican fragments. The serial mass spectrometry (ms/ms) allows the legion of beryllium to be sequenced and map back to the corresponding protein sequence. This is essential for understanding protein functions, post-translation modifications and interactions。
Protein-protein interaction measurement: the intersectional mass spectrometry (xl-ms) refers to the chemical interconnection of proteins at the point of interaction, and then to the mass spectrometry of the conjunctive compound. This can provide valuable information on protein-protein interface points and help model complex molecular structures。
Top-down proteomics: top-down proteomics refer to a direct mass spectrometry of the whole protein without prior digestion into a pelican section. This approach allows for detailed studies of protein isomers, post-translation modifications and higher structures. In particular, it applies to complex proteins such as manifestations of antibodies。
Hydrogen-thylene exchange mass spectrometry (hdx-ms): hdx-ms studies protein folding and kinetics by measuring the exchange of hydrogen atoms in the main protein chain with the atoms in the solvent. By comparing exchange rates in different areas of the protein, scientists can extrapolate information on the second and third levels of the protein structure。
Natural mass spectrometry: natural mass spectrometry can analyse biological molecules in natural, non-variant state. The technology is used to study large protein complexes, nucleic acids and membrane proteins in functional states, thus providing insight into their structure, chemical measurements and interactions。
In sum, mass spectrometry is a widely used and powerful analytical tool that has evolved over the past century into an important instrument in modern science, particularly in the field of structural biology. It is capable of ionizing, separating and testing the ion according to the mass ratio, thus allowing detailed studies of complex biological molecules with immeasurable value in applications such as protein identification, representation and interaction studies. When combined with separation techniques such as gas chromatography and liquid chromatography, mass spectrometry can provide incomparable resolution and accuracy in molecular structure analysis, thus providing insight into control mechanisms for understanding biological processes。
V. Common questions and answers
1. How many orders of magnitude can the difference between low and high
The main difference is the accuracy of the ion mass measured, with a low resolution that can only reach one integer or one integer, and a high resolution that can reach two or six decimal places; the high-resolution mass spectra can distinguish between two substances with very small differences in molecular mass, so that molecular elements can be accurately extrapolated。
2. Why do mass spectrometry models have positive and negative ion scans? How
The positive and negative ion patterns are two scanning models of mass spectrometry, with samples having both positive and negative charge ions following the ionization of esi sources, and some products having positive charge or negative charge depending on the physicochemical nature of the substance。
The selection of the model depends on whether the structure contains a matrix of stand-alone electron-friendly protons, e. G. -nh2, -oh (positive ion pattern), or a base of prone loss of protons, e. G. -cooh (positive ion model), some compounds can be measured as positive ion, e. G. Nucleotide。
Can mass spectrometry be used to test inorganic matter
Mass spectrometry is generally used for the identification analysis of organic molecules, and inorganics are recommended for analysis by means of icp-ms, ion chromatography (ic), chemical dripping, etc。
What are the ion sources of the mass spectra? How do we choose
Esi, apci, optical ionization sources of atmospheric pressure (appi), ei, chemical ionization sources (ci), base-based auxiliary laser ionization sources (maldi). Esi, apci, ei, ei are commonly used as soft ion, which largely does not produce debris peaks and is suitable for the analysis of polarized large molecular organic compounds; the apci source produces quasi-molecular ion peaks, with little debris generation, which is used mainly to analyse small molecular compounds of moderate or low polarity; and the ei is hard ion, which produces rich debris ion (but the molecular ion peaks are weak and difficult to capture), which is used mainly in volatile samples。
5. Why did the test result not show a mass weight peak of the expected molecular mass
This may be due to the inappropriate selection of the ion source or to the fact that the sample is too low to be detected。
6. Why does it need to be provided with the expected molecular volume and the molecular formula of the ingredients to be measured
The test is preceded by input into the molecular system of the ingredients to be detected, and the test results are automatically matched and the test is completed if the first test matches。



