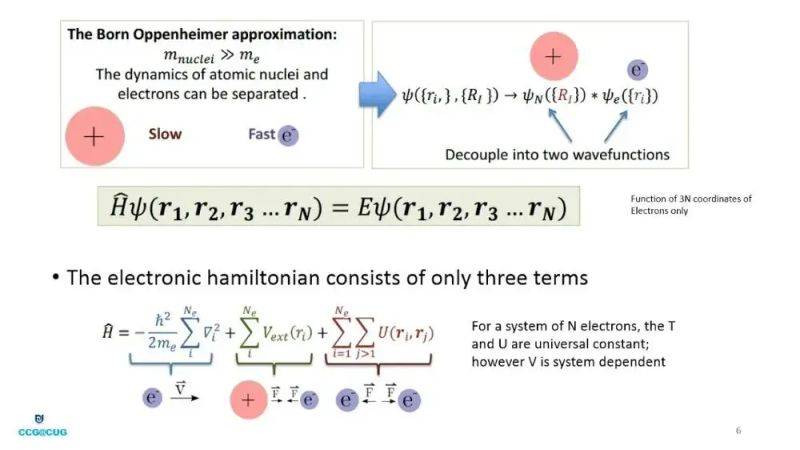
Note: density expresso theoryNal theory, dft is a quantum mechanics method used to solve the base-state nature of a multielectronic system. Its core idea is to describe the system by using electronic density rather than complex multi-electronic wave functions, to prove the energy of the only determining system of electronic density through the hohenberg-kohn theorem, and to simplify the complexity of calculation by transforming the multi-body problem into the movement of non-interactive electronics in an effective position。
Dft is widely used in materials science, catalytic, condensate physics and chemistry, and can accurately predict key properties such as structure, energy bands and adsorption energy。
Definition of the density general communication theory
Density expressoNal theory, dft, a calculation based on quantum mechanics, is used to study the base-state nature of a multielectronic system, whose core idea is to describe the physical properties of the system by using the electronic density distribution function (r) rather than the wave function。
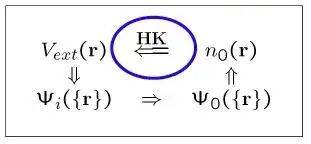
This theory was laid in 1964 by hohenberg and kohn, who demonstrated that there was a correspondence between the system's base-state electron density and the external field (e. G., the nuclear-generated force field) and that the system's energy could be expressed as a generic e-density, the smallest of which corresponded to the base-state energy. This revolutionary idea has reduced the complexity of calculations significantly by reducing the complex multi-body problem to a single particle problem。
Theory foundation: hohenberg-kohn theorem and kohn-sham equation
Hohenberg-kohn theorem contains two core elements:
Theorem of uniqueness: the only certainty of the outer field, vext(r) (except for constants), is the only determination of the nature of all the matrices。
Degradation principle: for any test of old (r) density, the minimum value of the energy pancreas e is the base energy and the smallest point corresponds to the base density。
However, the theorem does not give the specific form of a kinetic general letter t. To that end, kohn and sham proposed the kohn-sham equation: by constructing a virtual “non-interactive system” with the same electronic density as the real system, but without interaction between the electronics. The single electron formula for the system is:
Its active force, veff(r)=vext(r)+vh(r)+vxc(r), consists of three parts:
Vext: external (e. G. Atomic nuclear cologne)
Vh: hartree (the classic cologne of electrons)
Vxc: exchange of correlations (including quantum exchange and association effects)。
The electron density is constructed from a single wave function:
The specialty of the kohn-sham equation is to compress all multi-body interactions into a general exchange association letter, exc, and vxc = δexc/old. The equation is to be solved by a symmetrical (scf) integrator: the initial guess-density calculation of the effective force of the equation seeks to update the density of the equation until energy is reduced (usually Δe-5ev)。
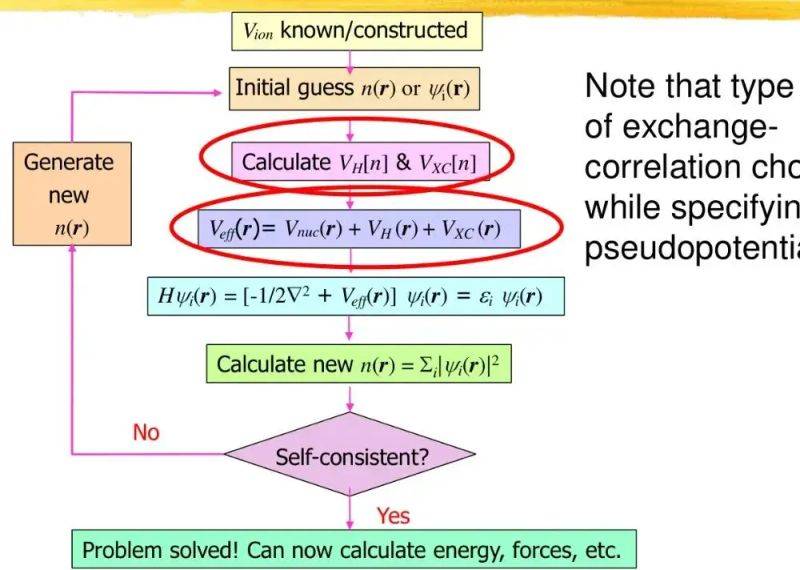
Approximation of the exchange of general communications: lda and gga
Since the exact form of exc is unknown, the actual calculation relies on the approximation model. The two most common approximations are:
Local density approximation (lda): assuming that exc at every point in space depends only on the old density (r) at that point, in the form of a flat electron gas model. Lda has a long time error of about 1 per cent, but seriously underestimates the combined energy (100 per cent error) and base height。
A broad gradient (gga) is an improvement of local density (lda) to better describe the non-equitable electronic system by introducing an electronic density gradient item (r)。
A typical gga general letter, such as pbe, can significantly improve the predictive accuracy of combined energy, atom structure and surface energy, reducing the combined energy error to about 5 per cent. However, gga usually stretches the chemical key slightly and understates the intensity of the constraint, indicating a “soft” trend in the structure of the system that requires attention in molecular and material simulations。
Dft applications in material science
Electronic structure and capacity analysis
In the case of silicon crystals, the dft calculates that the strip structure reveals a lacuna of approximately 1. 1ev and directly validates its semiconductor properties. The calculation process includes the construction of a diamond structure, the selection of gga-pbe general letters, the calculation of electron density in the self-concerting field, and the mapping of energy along high symmetric paths such as busy-x-l, which accurately shows the distribution of energy between the price belt and the belt。
Electronic density maps (e. G., examples of two-dimensional materials) visually show the charge space concentration: high density areas correspond to chemical keys or atomic core positions, and low density areas are gap areas。
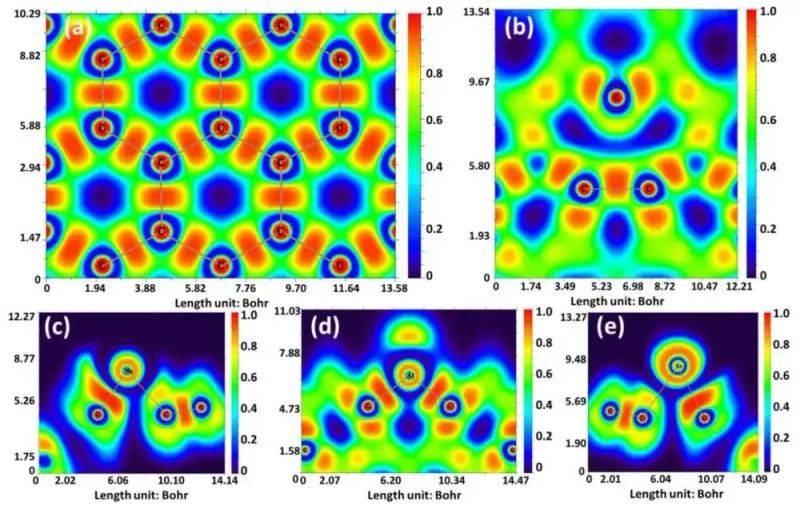
Doi: 10. 3390/s23125631
The enabling structure map is the core tool to reveal the electronic nature of the material, which intuitively reflects conductivity and lacuna size by showing the energy distribution of the electronic in the dynamic space。
For example, in two-dimensional materials, where the price belt is met at the same k point (e. G., a busy point) as the base of the conductor, the direct cleavage semiconductor is suitable for photoelectric applications; in the case of separation, the indirect zone material. The width of the bandwidth determines the energy required for the electron leap, which in turn affects the absorption of spectroscopy and streaming. It is possible to identify the conductor belts, the price to be taken, and the materials to be judged are metal, semiconductor or insulation。
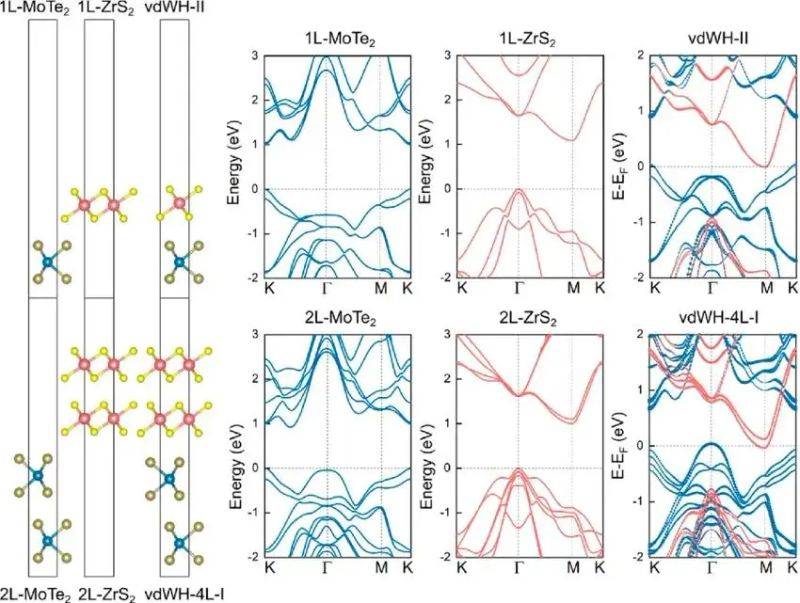
Doi: 10. 1021/acsami. 2c13151
Magnetic material and spin polarization
In melancholytic phosphorous systems (e. G., s), cyclic polarization calculations show a net magnetic rectangular of 0. 98 mb. Magnetic rectangular maps show that the pz orbit of the s atoms is swirling off with the py, pz orbit of the adjacent p atoms, and that the spin-up/down-state density (pdos) is asymmetrically near the pemi-energy level, resulting in the absence of a pair of electrons。
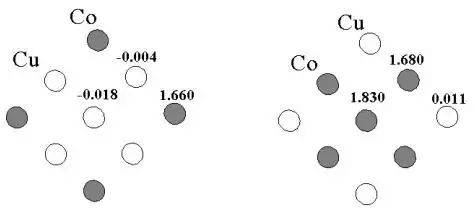
Similarly, the co-cu alloy magnetic rectangular map shows a surface of co-atomic rectangular up to 1. 66 mb, while the internal cu atomic magnetic rectangular is close to zero, revealing magneticity from the transition metal d electrons。
Differences in electron density between metal and insulation
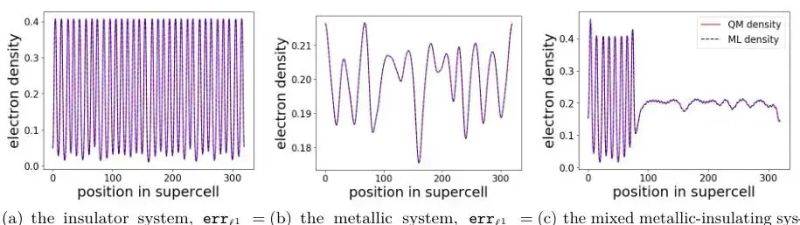
Three-dimensional electrodensity rendering clearly contrasts two systems:
Doi: 10. 1016/j. Jcp 2021. 110523
Metals: electron density space fluctuations are small and the phi-energy level is not zero, supporting continuous electron leaps
Insulation: density is cyclically oscillated, no electron state in the zone, and the charge localization is evident
Mixing systems: lan characteristics that combine metallic continuity and insulation。
Visualization of dft in chemical response machine studies
Path to transitional state search and response
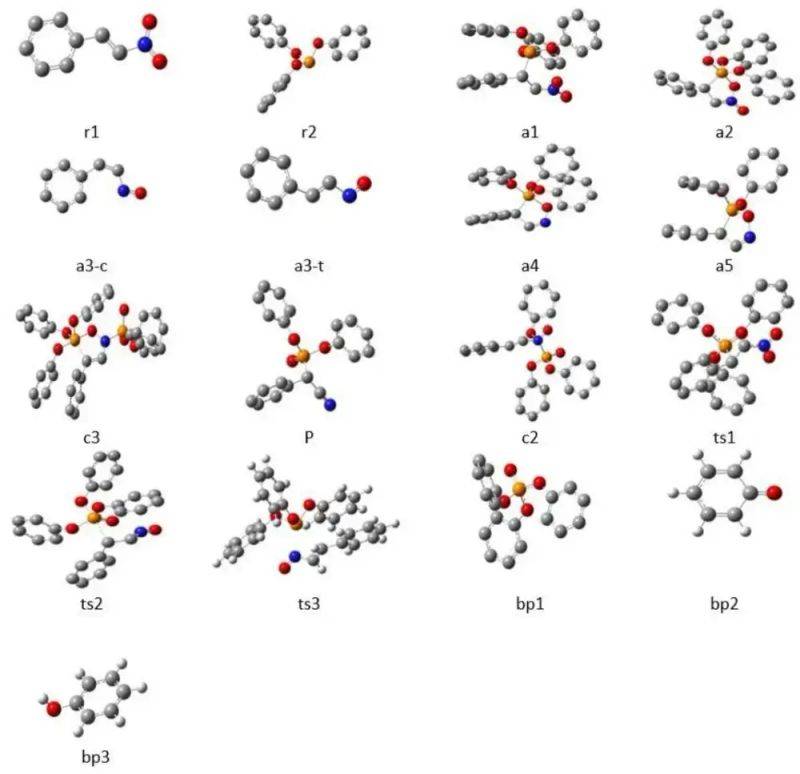
In the case of alpha-cyanate synthesis, for example, the response has four main phases: first, the reactional nitrostyrene and esters (r1, r2) present specific spatial configurations that provide a geometric basis for the response. It then enters a transition state (ts1, ts2), in the form of a change in the key length and the key angle, for example, the c-n key is pulled to about 1. 8 Å, while the nitrous vibration mode reveals the direction of the reaction coordinates and the reaction path。
An intermediate (a1, a3-c) is then formed for the sub-stable structure, with energy below the transitional state but still above the product, representing the structural state of a short presence. In the final product (bp1, bp2), the c-p co-priced key was successfully formed, and the nitro-commissioned service was reordered to cyanide to complete the reaction process。
Doi: 10. 21203/rs. 3. Rs-3737180/v1
Reaction energy profile
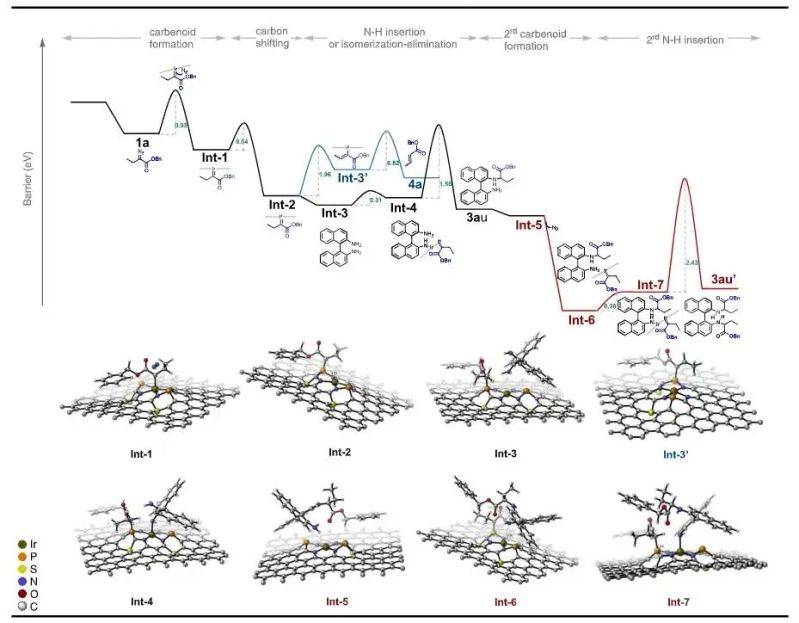
The reaction energy profile is based on the guibus free energy (ev) as a vertical axis and the reaction path as a cross-axis, which systematically depicts the transformation process from reaction to product. The changes in the free energy of the intermediates int-1 to int-7 reveal the reversibility and thermal stability of the response steps。
Among them, the key acceleration steps occurred in the transition phase of int-2 to ts1, with a capacity of up to 0. 8ev, with a decisive impact on the overall response rate. The high and low capacity reflects the level of stimulation required for this step and is one of the key parameters for optimizing the conditions for catalysts and reactions。
Doi: 10. 1021/acscatal. 3c05635
Limitations and directions for development
The density general communication theory (dft) is based on the hohenberg-kohn theorem and the kohn-sham equation and is widely applied to molecular and solid electronic structure simulations by simplifying the issue of multi-electronics to a single electronic force。
Dft, however, also faces a number of inherent challenges, including inaccurate descriptions of strong associated systems (such as high temperature superconductors and magnetic materials), inadequate handling of van der waals, and the general underestimation of 30-50 per cent by gga in semiconductor lacuna predictions。
To overcome these limitations, the current study is focusing on new approaches such as machine learning-aided general letter design, the optimization of general commodification and the integration of kinetic field theory (dmft). Evolving theories and algorithms are contributing to the continuous expansion of the capacity of the dft in catalytic, material design and energy research。